Use cases for coordinate mapping with ensembldb
Johannes Rainer, Laurent Gatto and Christian X. Weichenberger
Source:vignettes/coordinate-mapping-use-cases.Rmd
coordinate-mapping-use-cases.RmdThis documents describes two use cases for the coordinate system
mapping functionality of ensembldb: mapping of regions
within protein sequences to the genome and mapping of genomic to protein
sequence-relative coordinates. In addition, it showcases the advanced
filtering capabilities implemented in ensembldb.
Query for helix-loop-helix transcription factors on chromosome 21
Down syndrome is a genetic disorder characterized by the presence of all or parts of a third copy of chromosome 21. It is associated, among other, with characteristic facial features and mild to moderate intellectual disability. The phenotypes are most likely the result from a gene dosage-dependent increased expression of the genes encoded on chromosome 21 (Lana-Elola et al. 2011). Compared to other gene classes, transcription factors are more likely to have an immediate impact, even due to a moderate over-expression (which might be the result from gene duplication). One of the largest dimerizing transcription factor families is characterized by a basic helix-loop-helix domain (Massari and Murre 2000), a protein structural motif facilitating DNA binding.
The example below aims at identifying transcription factors with a
basic helix-loop-helix domain (Pfam ID PF00010) that are encoded on
chromosome 21. To this end we first load an R-library providing human
annotations from Ensembl release 86 and pass the loaded
EnsDb object along with a filter expression to the
genes method that retrieves the corresponding genes. Filter
expressions have to be written in the form
~ <field> <condition> <value> with
<field> representing the database column to be used
for the filter. Several such filter expressions can be concatenated with
standard R logical expressions (such as & or
|). To get a list of all available filters and their
corresponding fields, the supportedFilters(edb) function
could be used.
library(ensembldb)
library(EnsDb.Hsapiens.v86)
edb <- EnsDb.Hsapiens.v86
## Retrieve the genes
gns <- genes(edb, filter = ~ protein_domain_id == "PF00010" & seq_name == "21")The function returned a GRanges object with the genomic
position of the genes and additional gene-related annotations stored in
metadata columns.
gns## GRanges object with 3 ranges and 7 metadata columns:
## seqnames ranges strand | gene_id
## <Rle> <IRanges> <Rle> | <character>
## ENSG00000205927 21 33025845-33029196 + | ENSG00000205927
## ENSG00000184221 21 33070144-33072420 + | ENSG00000184221
## ENSG00000159263 21 36699133-36749917 + | ENSG00000159263
## gene_name gene_biotype seq_coord_system symbol
## <character> <character> <character> <character>
## ENSG00000205927 OLIG2 protein_coding chromosome OLIG2
## ENSG00000184221 OLIG1 protein_coding chromosome OLIG1
## ENSG00000159263 SIM2 protein_coding chromosome SIM2
## entrezid protein_domain_id
## <list> <character>
## ENSG00000205927 10215 PF00010
## ENSG00000184221 116448 PF00010
## ENSG00000159263 6493 PF00010
## -------
## seqinfo: 1 sequence from GRCh38 genomeThree transcription factors with a helix-loop-helix domain are
encoded on chromosome 21: SIM2, which is a master regulator of
neurogenesis and is thought to contribute to some specific phenotypes of
Down syndrome (Gardiner and Costa 2006)
and the two genes OLIG1 and OLIG2 for which genetic
triplication was shown to cause developmental brain defects (Chakrabarti et al. 2010). To visualize the
exonic regions encoding the helix-loop-helix domain of these genes we
next retrieve their transcript models and the positions of all Pfam
protein domains within the amino acid sequences of encoded by these
transcripts. We process SIM2 separately from OLIG1 and
OLIG2 because the latter are encoded in a narrow region on
chromosome 21 and can thus be visualized easily within the same plot. We
extract the transcript models for OLIG1 and OLIG2 that
encode the protein domain using the
getGeneRegionTrackForGviz function which returns the data
in a format that can be directly passed to functions from the
Gviz Bioconductor package (Hahne and
Ivanek 2016) for plotting. Since Gviz expects
UCSC-style chromosome names instead of the Ensembl chromosome names
(e.g. chr21 instead of 21), we change the
format in which chromosome names are returned by ensembldb
with the seqlevelsStyle method. All subsequent queries to
the EnsDb database will return chromosome names in UCSC
format.
## Change chromosome naming style to UCSC
seqlevelsStyle(edb) <- "UCSC"
## Retrieve the transcript models for OLIG1 and OLIG2 that encode the
## the protein domain
txs <- getGeneRegionTrackForGviz(
edb, filter = ~ genename %in% c("OLIG1", "OLIG2") &
protein_domain_id == "PF00010")Next we fetch the coordinates of all Pfam protein domains encoded by
these transcripts with the proteins method, asking for
columns "prot_dom_start", "prot_dom_end" and
"protein_domain_id" to be returned by the function. Note
that we restrict the results in addition to protein domains defined in
Pfam.
pdoms <- proteins(edb, filter = ~ tx_id %in% txs$transcript &
protein_domain_source == "pfam",
columns = c("protein_domain_id", "prot_dom_start",
"prot_dom_end"))
pdoms## DataFrame with 3 rows and 6 columns
## protein_domain_id prot_dom_start prot_dom_end protein_id tx_id
## <character> <integer> <integer> <character> <character>
## 1 PF00010 107 164 ENSP00000371785 ENST00000382348
## 2 PF00010 110 162 ENSP00000371794 ENST00000382357
## 3 PF00010 110 162 ENSP00000331040 ENST00000333337
## protein_domain_source
## <character>
## 1 pfam
## 2 pfam
## 3 pfamWe next map these protein-relative positions to the genome. We define
first an IRanges object with the coordinates and submit
this to the proteinToGenome function for mapping. Besides
coordinates, the function requires also the respective protein
identifiers which we supply as names.
pdoms_rng <- IRanges(start = pdoms$prot_dom_start, end = pdoms$prot_dom_end,
names = pdoms$protein_id)
pdoms_gnm <- proteinToGenome(pdoms_rng, edb)The result is a list of GRanges objects
with the genomic coordinates at which the protein domains are encoded,
one for each of the input protein domains. Additional information such
as the protein ID, the encoding transcript and the exons of the
respective transcript in which the domain is encoded are provided as
metadata columns.
pdoms_gnm## $ENSP00000371785
## GRanges object with 1 range and 7 metadata columns:
## seqnames ranges strand | protein_id tx_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr21 33070565-33070738 + | ENSP00000371785 ENST00000382348
## exon_id exon_rank cds_ok protein_start protein_end
## <character> <integer> <logical> <integer> <integer>
## [1] ENSE00001491811 1 TRUE 107 164
## -------
## seqinfo: 1 sequence from GRCh38 genome
##
## $ENSP00000371794
## GRanges object with 1 range and 7 metadata columns:
## seqnames ranges strand | protein_id tx_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr21 33027190-33027348 + | ENSP00000371794 ENST00000382357
## exon_id exon_rank cds_ok protein_start protein_end
## <character> <integer> <logical> <integer> <integer>
## [1] ENSE00001491833 2 TRUE 110 162
## -------
## seqinfo: 1 sequence from GRCh38 genome
##
## $ENSP00000331040
## GRanges object with 1 range and 7 metadata columns:
## seqnames ranges strand | protein_id tx_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr21 33027190-33027348 + | ENSP00000331040 ENST00000333337
## exon_id exon_rank cds_ok protein_start protein_end
## <character> <integer> <logical> <integer> <integer>
## [1] ENSE00001379164 1 TRUE 110 162
## -------
## seqinfo: 1 sequence from GRCh38 genomeColumn cds_ok in the result object indicates whether the
length of the CDS of the encoding transcript matches the length of the
protein sequence. For transcripts with unknown 3’ and/or 5’ CDS ends
these will differ. The mapping result has to be re-organized before
being plotted: Gviz expects a single GRanges
object, with specific metadata columns for the grouping of the
individual genomic regions. This is performed in the code block
below.
## Convert the list to a GRanges with grouping information
pdoms_gnm_grng <- unlist(GRangesList(pdoms_gnm))
pdoms_gnm_grng$id <- rep(pdoms$protein_domain_id, lengths(pdoms_gnm))
pdoms_gnm_grng$grp <- rep(1:nrow(pdoms), lengths(pdoms_gnm))
pdoms_gnm_grng## GRanges object with 3 ranges and 9 metadata columns:
## seqnames ranges strand | protein_id
## <Rle> <IRanges> <Rle> | <character>
## ENSP00000371785 chr21 33070565-33070738 + | ENSP00000371785
## ENSP00000371794 chr21 33027190-33027348 + | ENSP00000371794
## ENSP00000331040 chr21 33027190-33027348 + | ENSP00000331040
## tx_id exon_id exon_rank cds_ok
## <character> <character> <integer> <logical>
## ENSP00000371785 ENST00000382348 ENSE00001491811 1 TRUE
## ENSP00000371794 ENST00000382357 ENSE00001491833 2 TRUE
## ENSP00000331040 ENST00000333337 ENSE00001379164 1 TRUE
## protein_start protein_end id grp
## <integer> <integer> <character> <integer>
## ENSP00000371785 107 164 PF00010 1
## ENSP00000371794 110 162 PF00010 2
## ENSP00000331040 110 162 PF00010 3
## -------
## seqinfo: 1 sequence from GRCh38 genomeWe next define the individual tracks we want to visualize and plot
them with the plotTracks function from the
Gviz package.
library(Gviz)
## Define the individual tracks:
## - Ideogram
## ideo_track <- IdeogramTrack(genome = "hg38", chromosome = "chr21")
## - Genome axis
gaxis_track <- GenomeAxisTrack()
## - Transcripts
gene_track <- GeneRegionTrack(txs, showId = TRUE, just.group = "right",
name = "", geneSymbol = TRUE, size = 0.5)
## - Protein domains
pdom_track <- AnnotationTrack(pdoms_gnm_grng, group = pdoms_gnm_grng$grp,
id = pdoms_gnm_grng$id, groupAnnotation = "id",
just.group = "right", shape = "box",
name = "Protein domains", size = 0.5)
## Generate the plot
plotTracks(list(gaxis_track, gene_track, pdom_track))
Transcripts of genes OLIG1 and OLIG2 encoding a helix-loop-helix protein domain. Shown are all transcripts of the genes OLIG1 and OLIG2 that encode a protein with a helix-loop-helix protein domain (PF00010). Genomic positions encoding protein domains defined in Pfam are shown in light blue.
All transcripts are relatively short with the full coding region being in a single exon. Also, both transcripts encode a protein with a single protein domain, the helix-loop-helix domain PF00010.
Next we repeat the analysis for SIM2 by first fetching all of its transcript variants encoding the PF00010 Pfam protein domain from the database. Subsequently we retrieve all Pfam protein domains encoded in these transcripts.
## Fetch all SIM2 transcripts encoding PF00010
txs <- getGeneRegionTrackForGviz(edb, filter = ~ genename == "SIM2" &
protein_domain_id == "PF00010")
## Fetch all Pfam protein domains within these transcripts
pdoms <- proteins(edb, filter = ~ tx_id %in% txs$transcript &
protein_domain_source == "pfam",
columns = c("protein_domain_id", "prot_dom_start",
"prot_dom_end"))At last we have to map the protein domain coordinates to the genome and prepare the data for the plot. Since the code is essentially identical to the one for OLIG1 and OLIG2 it is not displayed.
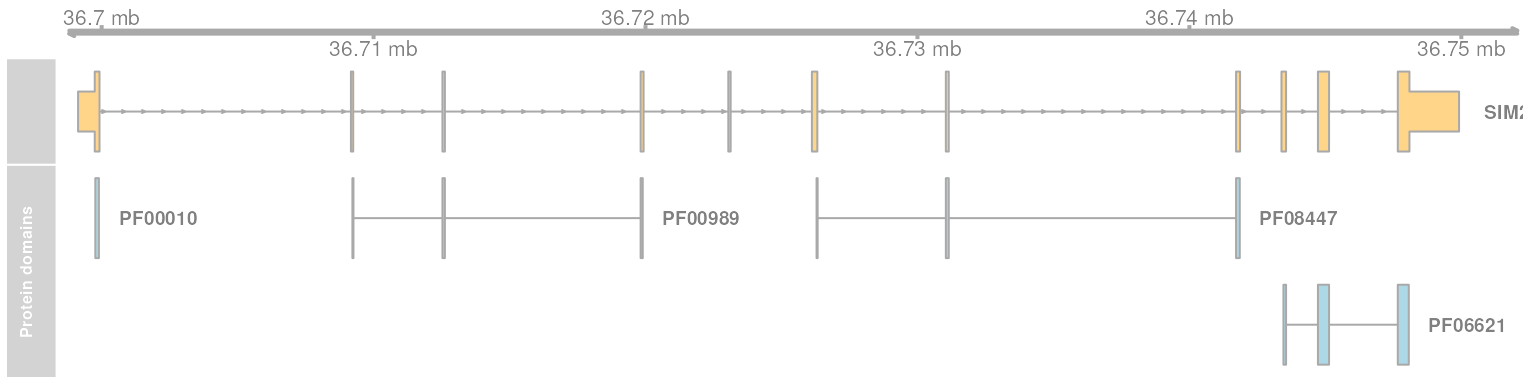
Transcripts of the gene SIM2 encoding a helix-loop-helix domain. Shown are all transcripts of SIM2 encoding a protein with a helix-loop-helix protein domain (PF00010). Genomic positions encoding protein domains defined in Pfam are shown in light blue.
The SIM2 transcript encodes a protein with in total 4 protein domains. The helix-loop-helix domain PF00010 is encoded in its first exon.
Mapping of genomic coordinates to protein-relative positions
One of the known mutations for human red hair color is located at
position 16:89920138 (dbSNP ID rs1805009) on the human genome (version
GRCh38). Below we map this genomic coordinate to the respective
coordinate within the protein sequence encoded at that location using
the genomeToProtein function. Note that we use
"chr16" as the name of the chromosome, since we changed the
chromosome naming style to UCSC in the previous example.
gnm_pos <- GRanges("chr16", IRanges(89920138, width = 1))
prt_pos <- genomeToProtein(gnm_pos, edb)
prt_pos## IRangesList object of length 1:
## [[1]]
## IRanges object with 3 ranges and 8 metadata columns:
## start end width | tx_id cds_ok
## <integer> <integer> <integer> | <character> <logical>
## ENSP00000451605 294 294 1 | ENST00000555147 TRUE
## ENSP00000451760 294 294 1 | ENST00000555427 TRUE
## ENSP00000451560 294 294 1 | ENST00000556922 TRUE
## exon_id exon_rank seq_start seq_end seq_name
## <character> <integer> <integer> <integer> <character>
## ENSP00000451605 ENSE00002458332 1 89920138 89920138 chr16
## ENSP00000451760 ENSE00002477640 3 89920138 89920138 chr16
## ENSP00000451560 ENSE00002507842 1 89920138 89920138 chr16
## seq_strand
## <character>
## ENSP00000451605 *
## ENSP00000451760 *
## ENSP00000451560 *The genomic position could thus be mapped to the amino acid 294 in
each of the 3 proteins listed above. Using the select
function we retrieve the official symbol of the gene for these 3
proteins.
## SYMBOL PROTEINID
## 1 RP11-566K11.2 ENSP00000451560
## 2 MC1R ENSP00000451605
## 3 MC1R ENSP00000451760Two proteins are from the MC1R gene and one from RP11-566K11.2 (ENSG00000198211) a gene which exons overlap exons from MC1R as well as exons of the more downstream located gene TUBB3. To visualize this we first fetch transcripts overlapping the genomic position of interest and subsequently all additional transcripts within the region defined by the most downstream and upstream exons of the transcripts.
## Get transcripts overlapping the genomic position.
txs <- getGeneRegionTrackForGviz(edb, filter = GRangesFilter(gnm_pos))
## Get all transcripts within the region from the start of the most 5'
## and end of the most 3' exon.
all_txs <- getGeneRegionTrackForGviz(
edb, filter = GRangesFilter(range(txs), type = "within"))
## Plot the data
## - Ideogram
## ideo_track <- IdeogramTrack(genome = "hg38", chromosome = "chr16")
## - Genome axis
gaxis_track <- GenomeAxisTrack()
## - Transcripts
gene_track <- GeneRegionTrack(all_txs, showId = TRUE, just.group = "right",
name = "", geneSymbol = TRUE, size = 0.5)
## - highlight the region.
hl_track <- HighlightTrack(list(gaxis_track, gene_track), range = gnm_pos)
## Generate the plot
plotTracks(list(hl_track))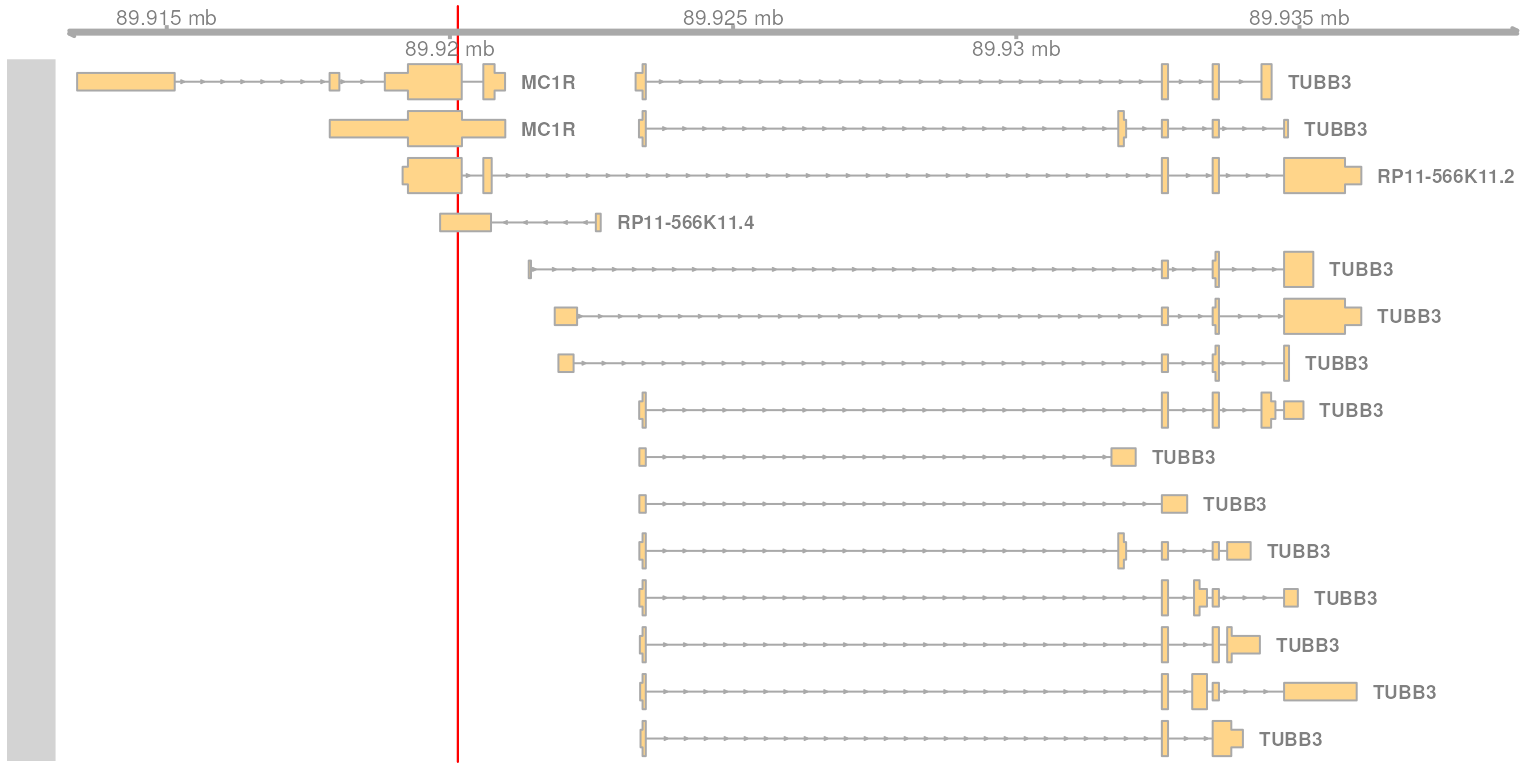
Transcripts overlapping, or close to, the genomic position of interest. Shown are all transcripts The genomic position of the variant is highlighted in red.
In the plot above we see 4 transcripts for which one exon overlaps
the genomic position of the variant: two of the gene MC1R, one
of RP11-566K11.2 and one of RP11-566K11.4, a
non-coding gene encoded on the reverse strand. Using the
proteins method we next extract the sequences of the
proteins encoded by the 3 transcripts on the forward strand and
determine the amino acid at position 294 in these. To retrieve the
results in a format most suitable for the representation of amino acid
sequences we specify return.type = "AAStringSet" in the
proteins call.
## Get the amino acid sequences for the 3 transcripts
prt_seq <- proteins(edb, return.type = "AAStringSet",
filter = ~ protein_id == names(prt_pos[[1]]))
## Extract the amino acid at position 294
library(Biostrings)
subseq(prt_seq, start = 294, end = 294)## AAStringSet object of length 3:
## width seq names
## [1] 1 D ENSP00000451560
## [2] 1 D ENSP00000451760
## [3] 1 D ENSP00000451605The amino acid at position 294 is for all an aspartic acid (“D”) which is in agreement with the reference amino acid of mutation Asp294His (Valverde et al. 1995) described by the dbSNP ID of this example.
Session information
## R Under development (unstable) (2023-02-16 r83857)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.1 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] grid stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] Biostrings_2.67.0 XVector_0.39.0
## [3] Gviz_1.43.1 EnsDb.Hsapiens.v86_2.99.0
## [5] ensembldb_2.23.2 AnnotationFilter_1.23.0
## [7] GenomicFeatures_1.51.4 AnnotationDbi_1.61.0
## [9] Biobase_2.59.0 GenomicRanges_1.51.4
## [11] GenomeInfoDb_1.35.15 IRanges_2.33.0
## [13] S4Vectors_0.37.3 BiocGenerics_0.45.0
## [15] BiocStyle_2.27.1
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 rstudioapi_0.14
## [3] jsonlite_1.8.4 magrittr_2.0.3
## [5] rmarkdown_2.20 fs_1.6.1
## [7] BiocIO_1.9.2 zlibbioc_1.45.0
## [9] ragg_1.2.5 vctrs_0.5.2
## [11] memoise_2.0.1 Rsamtools_2.15.1
## [13] RCurl_1.98-1.10 base64enc_0.1-3
## [15] htmltools_0.5.4 progress_1.2.2
## [17] curl_5.0.0 Formula_1.2-4
## [19] sass_0.4.5 bslib_0.4.2
## [21] htmlwidgets_1.6.1 desc_1.4.2
## [23] cachem_1.0.6 GenomicAlignments_1.35.0
## [25] lifecycle_1.0.3 pkgconfig_2.0.3
## [27] Matrix_1.5-3 R6_2.5.1
## [29] fastmap_1.1.0 GenomeInfoDbData_1.2.9
## [31] MatrixGenerics_1.11.0 digest_0.6.31
## [33] colorspace_2.1-0 rprojroot_2.0.3
## [35] textshaping_0.3.6 Hmisc_4.8-0
## [37] RSQLite_2.3.0 filelock_1.0.2
## [39] fansi_1.0.4 httr_1.4.4
## [41] compiler_4.3.0 bit64_4.0.5
## [43] htmlTable_2.4.1 backports_1.4.1
## [45] BiocParallel_1.33.9 DBI_1.1.3
## [47] highr_0.10 biomaRt_2.55.0
## [49] rappdirs_0.3.3 DelayedArray_0.25.0
## [51] rjson_0.2.21 tools_4.3.0
## [53] foreign_0.8-84 nnet_7.3-18
## [55] glue_1.6.2 restfulr_0.0.15
## [57] checkmate_2.1.0 cluster_2.1.4
## [59] generics_0.1.3 gtable_0.3.1
## [61] BSgenome_1.67.4 data.table_1.14.8
## [63] hms_1.1.2 xml2_1.3.3
## [65] utf8_1.2.3 pillar_1.8.1
## [67] stringr_1.5.0 splines_4.3.0
## [69] dplyr_1.1.0 BiocFileCache_2.7.2
## [71] lattice_0.20-45 survival_3.5-3
## [73] rtracklayer_1.59.1 bit_4.0.5
## [75] deldir_1.0-6 biovizBase_1.47.0
## [77] tidyselect_1.2.0 knitr_1.42
## [79] gridExtra_2.3 bookdown_0.32
## [81] ProtGenerics_1.31.0 SummarizedExperiment_1.29.1
## [83] xfun_0.37 matrixStats_0.63.0
## [85] stringi_1.7.12 lazyeval_0.2.2
## [87] yaml_2.3.7 evaluate_0.20
## [89] codetools_0.2-19 interp_1.1-3
## [91] tibble_3.1.8 BiocManager_1.30.19
## [93] cli_3.6.0 rpart_4.1.19
## [95] systemfonts_1.0.4 munsell_0.5.0
## [97] jquerylib_0.1.4 dichromat_2.0-0.1
## [99] Rcpp_1.0.10 dbplyr_2.3.0
## [101] png_0.1-8 XML_3.99-0.13
## [103] parallel_4.3.0 ellipsis_0.3.2
## [105] pkgdown_2.0.7.9000 ggplot2_3.4.1
## [107] assertthat_0.2.1 blob_1.2.3
## [109] prettyunits_1.1.1 latticeExtra_0.6-30
## [111] jpeg_0.1-10 bitops_1.0-7
## [113] VariantAnnotation_1.45.1 scales_1.2.1
## [115] purrr_1.0.1 crayon_1.5.2
## [117] rlang_1.0.6 KEGGREST_1.39.0